

How To Install Gromacs On Windows
Compiling and running GROMACS natively on Windows
I have been working for a few months on a project on molecular dynamics (MD) simulations. I was non particularly familiar with MD simulations before this, but I had to learn to use NAMD and GROMACS — ii very popular softwares that can run various type of calculations using force-fields (i.e. the atoms are considered as classical particles obeying Newtonian dynamics and the forces between them are expressed by parameterised equations). In that location are other softwares for Dr. simulations, such as OpenMM, only my guess is that GROMACS and NAMD claim the king of beasts's share of papers published well-nigh MD.
NAMD provides precompiled binaries for Windows, just GROMACS does not, and so information technology needs to be compiled. On Linux or POSIX-like systems compiling is quite easy. On Windows, information technology can be compiled, just the procedure is a bit difficult.
TL, DR: If you lot just want the compiled binaries, and do not desire to read further, and then please visit this github repository where I have put precompiled Windows binaries of GROMACS.
Here'due south how to compile GROMACS for Windows natively (i.e. no Cygwin or WSL):
Requirements —
- Windows x (Windows 8 and 7 should besides piece of work simply haven't been tested)
- Microsoft Visual Studio 2019 (2017 also works, older versions may work)
- [Intel oneAPI base toolkit (for MKL)] OR [FFTW3 library]
- (optional) meson and ninja
Installing Visual Studio 2019
The community edition of Visual Studio 2019 (or 2017) can exist downloaded for gratis from Microsoft website. The community edition is sufficient for our purpose. (Download page: latest version(VS2019) , VS2017 and older)
Download and run the installer. When the installer asks for which components should exist installed, make certain to select "Desktop evolution with C++". The other options are not necessary for installing GROMACS, so they can be left unchecked if you lot wish.
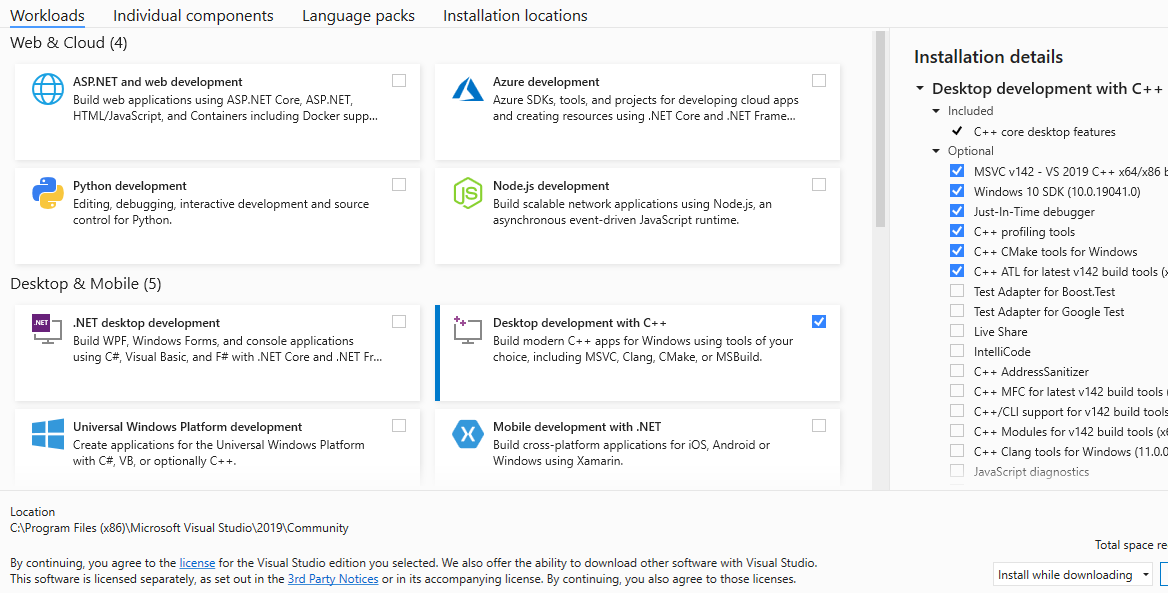
I recommend not changing any of the optional installations on the right side. The "C++ CMake tools for Windows" are required for building GROMACS. Click Install to download and install Visual Studio.
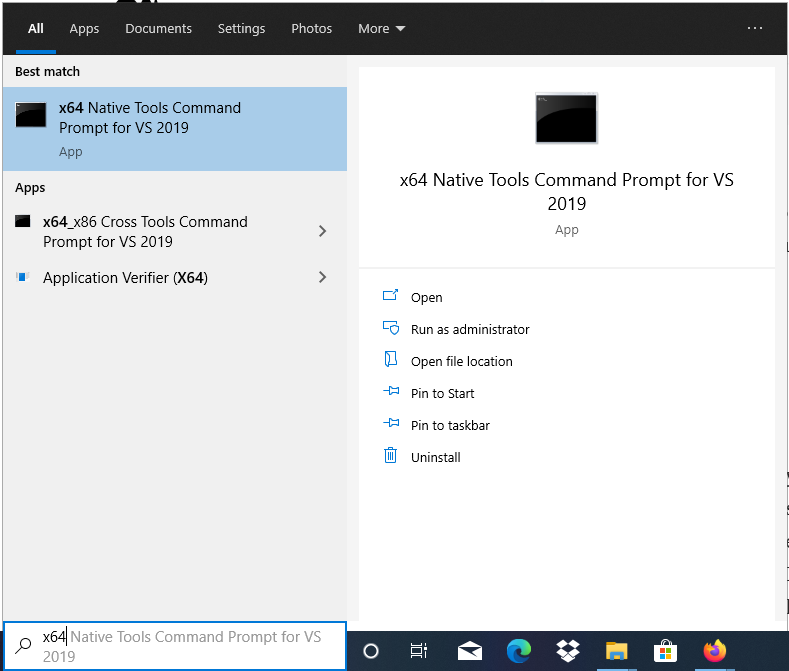
We will apply the command line compilation. For this the compiler control line surround has to exist brought up. Please cheque that yous have the option "x64 Native Tools Command Prompt for VS2019" from the get-go menu like this:
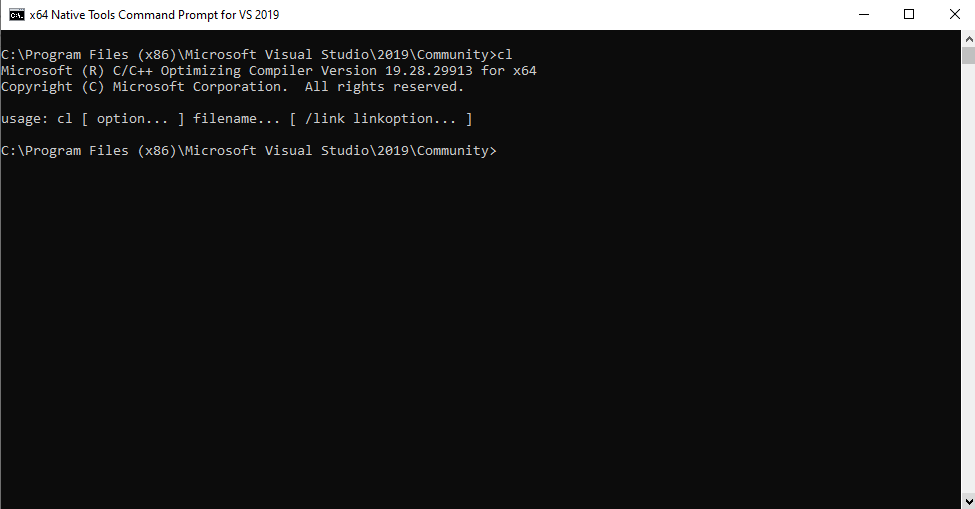
Open the compiler command prompt and bank check that running cl gives an output like this: (cl.exe is the MSVC++ compiler)
Installing Intel oneAPI for MKL (alternative to FFTW3)
GROMACS needs fast fourier transform libraries for many simulation options, such as particle mesh ewald electrostatics. These libraries tin can be provided by Intel MKL (Math Kernel Library). Intel MKL is quite fast, especially on Intel chips. (I am not sure if it is equally fast on other fries like AMD; there were reports some time ago most Intel inserting code into MKL that checks if the processor is Intel, and then runs the slower code if it isn't. Merely in the 2020 update 3, Intel claims to have stock-still them.)
If you lot cull MKL for GROMACS, MKL will also exist used for linear algebra (BLAS and LAPACK). Intel MKL is now freely available as a office of the Intel oneAPI base toolkit (Download page: base toolkit). Select Windows as Operating system. I would recommend downloading the online installer, every bit the local installer has the full oneAPI base of operations kit which is not necessary. And so click download, the website volition prompt you to sign in — click download equally guest if yous don't have an account.
Run the installer once downloaded. Make sure to select only the "Intel oneAPI Math Kernel Library" option because the others are not required.
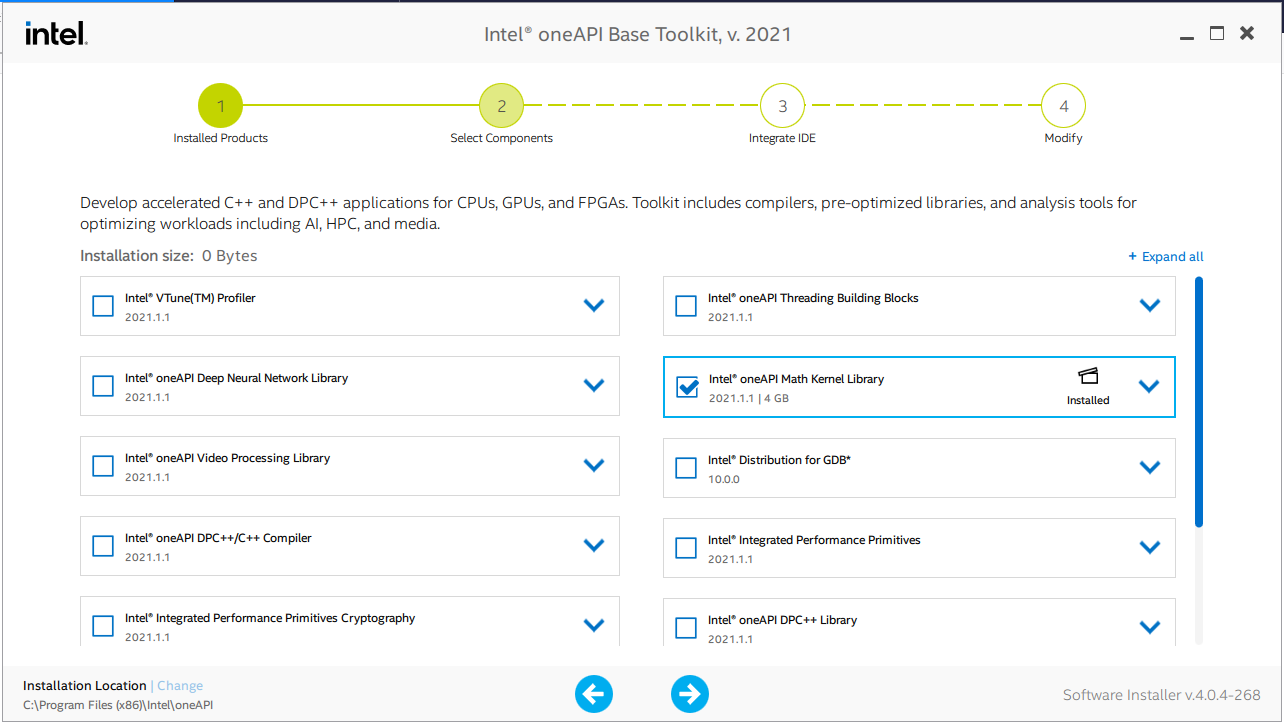
Note the installation location at the bottom left corner. Make sure that you can detect the libraries mkl_intel_lp64.lib, mkl_sequential.liband mkl_core.lib from the installation directory. If the default is kept, then those libraries should be in C:\Program Files (x86)\Intel\oneAPI\mkl\latest\lib\intel64\ .
Using FFTW3 library (alternative to MKL)
If you lot are using Intel fries, MKL will give yous the best performance. MKL is also works on AMD chips, but information technology is possible that other libraries volition give faster performance in that location. You lot would accept to run benchmarks and check for yourself. FFTW3 is one such open up-source library which provides fast fourier transform functionality.
To use FFTW3 download the library from https://www.fftw.org/install/windows.html. Unfortunately, FFTW3 provides source code by default and no official windows precompiled libraries. The page linked provides libraries for windows that take been compiled by other users. Note that the windows libraries are of an older version (3.3.5) not the latest (3.3.ix). If you wish to use FFTW3, download the 64-bit version zip and extract into a binder.
For the usual build of GROMACS, the unmarried precision FFTW3 library is required. Every bit MSVC++ compiler is being used here, the library needs to exist nowadays in import library format (.lib), which is not provided in the archive.
To brand the .lib files, open the compiler command prompt for MSVC++ ("x64 Native Tools …"). Navigate to the folder where the FFTW3 files are present, so run lib /machine:x64 /def:libfftw3f-3.def . This will generate a libfftw3f-f.lib file. This is the single precision FFTW3 import library that we will need while compiling GROMACS.
(Optional) Meson and Ninja
Ninja is a build system that tin can take build files generated from CMake and bear out the compiling and linking to prepare the final executable software. Ninja and meson are usually distributed together for Windows in an installer. (we really don't accept any need for meson)
Do we demand to apply ninja for building GROMACS? No, because Visual Studio already has a build arrangement (nmake) which can do the same job, and is installed with Visual Studio by default. However, ninja tin can employ all of the cores of your PC to run parallel compile jobs, whereas nmake but runs compilations sequentially, so it takes a lilliputian longer. For the purposes of this, the fourth dimension difference isn't much, so you can utilize nmake or ninja , whichever you lot wish.
Downloading GROMACS
GROMACS releases source codes at very frequent intervals. The source codes of different versions can be downloaded from this page. As they change the source code slightly in each version, it is possible that futurity versions may cause errors during compiling. I am using the 2021.1 release for this guide.
Download the tar.gz archive, and then excerpt it into a folder.
Some modifications
For some versions, modifications might exist required.
For the 2021.ane version, one filename needs to be changed. Become to the folder where the extracted source files are, and discover the file src\gromacs\nbnxm\kernels_simd_2xmm\kernel_ElecQSTabTwinCut_VdwLJEwCombGeom_VgrpF.cpp0000666. The extension of this file should be just .cpp, so remove those numbers. (I am not really sure why the file has a weird extension, but without the file, I am getting Unresolved Externals error during linking.) This problem is not present in 2021.2 or more recent versions.
For the 2021 version, no changes are required. I believe the aforementioned applies for older versions.
Running CMake to generate build files
CMake is arranged with Visual Studio and can exist run from the x64 Native Tools command prompt. From the compiler command prompt, navigate to the folder where GROMACS source files have been extracted.
Run the post-obit commands in order (if using Intel MKL):
set CC=cl
ready CXX=cl cmake -Yard"NMake Makefiles" -Due south. -B./build -DGMX_GPU=off -DGMX_FFT_LIBRARY=mkl -DCMAKE_BUILD_TYPE=Release -DBUILD_SHARED_LIBS=off -DMKL_INCLUDE_DIR="C:/Program Files (x86)/Intel/oneAPI/mkl/latest/include" -DMKL_LIBRARIES="C:/Program Files (x86)/Intel/oneAPI/mkl/latest/lib/intel64/mkl_intel_lp64.lib;C:/Programme Files (x86)/Intel/oneAPI/mkl/latest/lib/intel64/mkl_sequential.lib;C:/Plan Files (x86)/Intel/oneAPI/mkl/latest/lib/intel64/mkl_core.lib"
The outset few lines prepare a few environment variables (which are non always necessary, but I am setting them just to exist condom). The CC and CXX variables store the proper noun of the C and C++ compiler that CMake will use respectively (here MSVC++ is used for both).
(Notation: the cmake control is one whole command i.e. not broken into lines)
In that location are several things to discover here. The -Yard pick determines to build system used for compilation. Here the argument "NMake Makefiles" specifies nmake. If you wish to apply Ninja, utilise the statement Ninja (without quotes i.e. -GNinja ). The -S option tells cmake that the current directory (.) is the source directory. The -B option tells it to create a binder ./build and place all the configured build files in that location. -DGMX_GPU=off means GPU acceleration is not used (every bit I do not currently have a GPU; if I get one, I will update this with a GPU accelerated build guide).
And so the DGMX_FFT_LIBRARY=mkl chooses Intel MKL equally the library for Fourier-transform. -DCMAKE_BUILD_TYPE=Release means highest optimization is used, without whatsoever debugging facility.-DBUILD_SHARED_LIBS=off ways the GROMACS binary is statically linked to the gromacs library (gromacs.lib) and is more portable.
Every bit we have specified the Intel MKL as the fast Fourier-transform library, we demand to specify its location. The -DMKL_INLCUDE_DIR gives the location of the include files (e.g. headers such equally mkl.h). The default installation directory for Intel oneAPI is C:/Plan Files (x86)/Intel/oneAPI/mkl/latest/include . If you installed it in a unlike location, make sure to change the path accordingly.
The -DMKL_LIBRARIES gives the location of the libraries. Three libraries are required for MKL static link: mkl_intel_lp64.lib , mkl_sequential.lib and mkl_core.lib . The full paths to each of these libraries accept to given, separated by a semicolon. Equally before, if you lot installed oneAPI in a different location, change accordingly.
Note that each path statement has to be inside a pair of double quotes (" " ) because they contain spaces. Also find the use of frontslash (/) in the paths instead of the Windows mode backslash (\). This is considering CMake takes paths with frontslashes, as information technology treats backslash as escape character. You lot must utilise frontslash, otherwise CMake won't be able to find the files.
After you run this command, CMake will test out a lot of different options and will endeavor to choose the best possible compilation options on your PC. Don't worry if there are a few " — Failed" messages. At the end, information technology should show you the message "Generating washed" and "Build files have been written to…". This ways CMake configuration is successful.
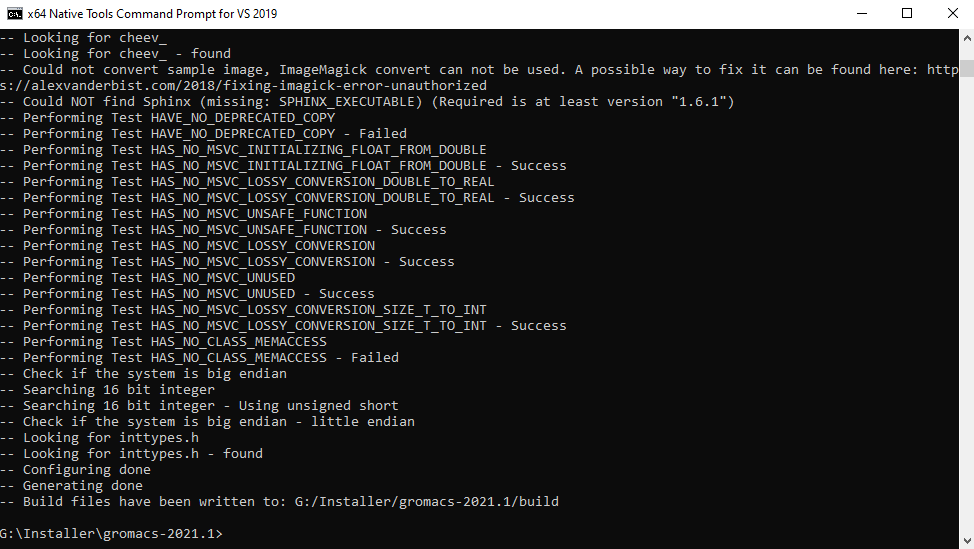
When using FFTW3 library, run these commands:
set CC=cl
set CXX=cl cmake -G"Nmake Makefiles" -Due south. -B./build -DGMX_GPU=off -DGMX_FFT_LIBRARY=fftw3 -DCMAKE_BUILD_TYPE=Release -DBUILD_SHARED_LIBS=off -DFFTWF_INCLUDE_DIR="G:/Installer/gromacs-2021.1/fftw/fftw-3.3.5-dll64" -DFFTWF_LIBRARY="Yard:/Installer/gromacs-2021.1/fftw/fftw-3.three.5-dll64/libfftw3f-iii.lib"
Here, the -DFFTWF_INCLUDE_DIR should point to the directory where the inlcude file fftw3.h is nowadays. The -DFFTWF_LIBRARY should signal to the library file for single precision FFTW3 (libfftw3f-3.lib)
Over again, CMake should generate the build files successfully.
Last compilation and linking
The final compilation and linking can exist washed either past nmake or ninja . NMake can be run from the x64 Native Tools command prompt. If you used the meson-ninja installer for Windows, so ninja is automatically added to the PATH during installation, then ninja tin can be run from whatever command prompt.
Navigate to the build folder in the GROMACS source folder, and and then run NMake:
cd build
nmake For Ninja, run:
cd build
ninja -j 4 Here, -j 4 ways Ninja will use 4 cores to run parallel compilation processes. If you have more cores in your chip, then change the value.
The compilation will take some fourth dimension (~20 minutes). You will see a long output, and there might be a few compiler warnings — they are ok to ignore. Unless there is whatever fault message, at the terminate you will see the message "Linking gmx.exe" or something similar to that. If the linking succeeds, the binary gmx.exe can be plant in the build folder.
If you used Ninja, the gmx.exe is more often than not in the build/bin folder. If you used NMake, the gmx.exe is probably in the build folder itself.
(Optional) MPI parallel builds
The default build of GROMACS tin apply OpenMP parallelisation, and/or it's own thread-based parallelisation (called thread-MPI). Both are quite efficient. Yet, if there are a very high number of processors, or you lot are running it on multiple processors (nodes) and so MPI-parallelisation is better. It is unlikely that yous will get whatsoever advantage with MPI based builds on a laptop or abode desktop. Nevertheless, if y'all want to utilise a MPI parallelised GROMACS binary, first install MS-MPI from here. Then add the argument DGMX_MPI=on to the CMake invocation command. CMake will automatically find the location of MS-MPI and compile with it.
Notation that turning on MPI will automatically turn off thread-MPI. You volition all the same take OpenMP support. Likewise note that the exectuable will be named gmx_mpi.exe later on compilation finishes.
Setting up GROMACS for run
Outset, choose a directory where you will keep GROMACS for running. For example, you could keep it in "C:\gromacs". Brand sure that the directory proper name hasn't got whatever spaces in it.
Create a binder "bin" within this binder, and then put gmx.exe insider the "bin" sub-binder.
Then go to the binder where y'all extracted the gromacs source files. Find the binder "share". Copy the "share" folder into your GROMACS directory. And then within the share folder create a new folder named "gromacs" and put the template and height directories into that. (Wait at the directory tree below if it's confusing).
GROMACS uses OpenMP parallelisation and the Microsoft Visual C/C++ runtime. These are provided by .dll files (dynamic link libraries). I am not entirely sure whether Visual Studio automatically installs them or non. If they are non installed, the program will not run. You tin also install only the dll files from the Visual C++ Redistributable Package hither.
If you used Intel MKL:
The GROMACS executable was linked statically to Intel MKL, so no dll is necessary.
If you used FFTW3:
Discover the libfftw3f-3.dll file in the directory where FFTW3 was extracted. Re-create this file into the "bin" sub-folder where gmx.exe is present.
(Optional) Copy the gromacs library
At that place should exist a gromacs.lib or gmx.lib library nowadays inside the "lib" folder insider build folder when y'all cease compilation. This is required just if yous are going to use GROMACS in another program. Yous tin copy these files to a "lib" sub-folder within the GROMACS install directory, but I don't believe they are necessary.
(Note: if you didn't use -DBUILD_SHARED_LIBS=off during compilation so a gromacs.dll or gmx.dll file tin can be created. You have to copy those files into the "bin" binder probably.)
At the end, your GROMACS directory tree should look like this:
C:\gromacs\
├───bin
│ ├───gmx.exe
│ ├───vcomp140.dll
│ └───[libfftw3f-three.dll] (just if you used FFTW3)
├───lib
│ └───gromacs.lib
│
└───share
└───gromacs
├───template
│ └───cmake
└───top
├───amber03.ff
├───amber94.ff
├───amber96.ff
├───amber99.ff
├───amber99sb-ildn.ff
├───amber99sb.ff
├───amberGS.ff
├───charmm27.ff
├───gromos43a1.ff
├───gromos43a2.ff
├───gromos45a3.ff
├───gromos53a5.ff
├───gromos53a6.ff
├───gromos54a7.ff
└───oplsaa.ff Once all the files are in identify, we demand to create a mode to access the GROMACS binary and those files from command line.
For this, create a file named "GMXRC.bat" with the following contents:
@set "PATH=C:\Gromacs\bin;%PATH%"
@set GMXDATA=C:\Gromacs\share\gromacs
@cmd Modify the paths if y'all are using a different location. Save this .bat file somewhere user-friendly.
Whenever, you need to run GROMACS, just double click the bat file and it volition open a command prompt window where yous can run GROMACS. Type gmx and press enter; you should run across something similar this:
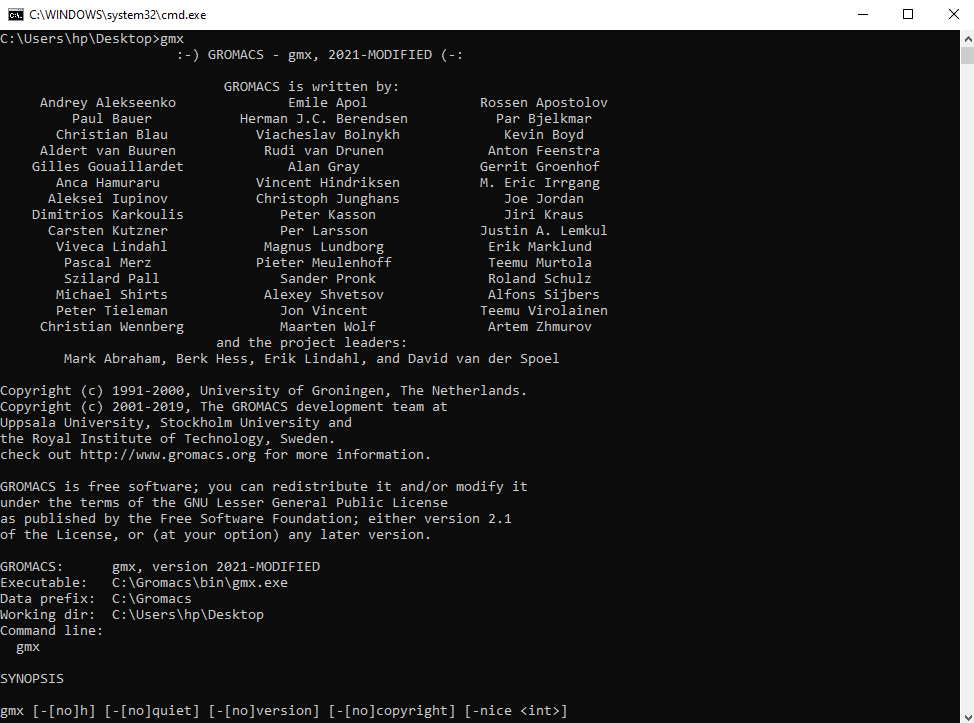
For MPI-parallel builds, you will accept to apply the MS-MPI mpiexec command to kick off the binary. And so, if you accept 4 processors on your computer you would use mpiexec -np four gmx_mpi.exe <commands> . The mpiexec command is automatically installed into your PATH when yous install MS-MPI.
Regression tests
You can test whether y'all compiled your GROMACS correctly by running regression tests. These examination can be downloaded from the same webpage as the GROMACS source codes (e.thou. for v2021.i download here).
You will also need Perl to run the tests. The tests do non accept long — at most 3 minutes. So, I would recommend running the tests in one case. If everything is right, then at the end y'all would meet the bulletin "All 7 essential dynamic tests PASSED".
If any of the tests failed, there was probably an error in compilation.
Well that's information technology! Yous take GROMACS up and running on your Windows PC.
Is information technology worth it?
Every bit you lot can see, compiling on Windows is quite a bit of pain, because y'all need to explicitly specify the paths to the libraries and include files etc.
Is information technology worth going through all of this problem? When getting into computational modelling I was regularly told "why don't you get Linux?" when faced with softwares that won't run on Windows. My laptop came with Windows loaded, and I have all of my files and utilities that I need regularly. It was too much of risk to attempt to install a dual boot Linux. Despite Linux beingness quite a robust and former Os, a lot of utilities nonetheless do not exist on Linux. For example, I demand to use a VPN for my academy work, and that VPN client only installs on Windows or Mac.
Admitted, for whatever major product calculations you will need to use a HPC (i.e. a supercomputer). But a lot of the testing and preparatory piece of work has to be washed on my own PC, and pocket-sized production runs can even be washed if your PC is strong enough. So I think it is worth it, to compile GROMACS or other softwares on Windows if they can run on it.
Many people mention Cygwin and WSL whenever compiling source code (targeted at Linux) on Windows is mentioned. But they are both a Linux compatibility layers, and very slow especially if file I/O is involved. Using them should be terminal resort, non the first option.
As mentioned at the first I have also placed my compiled GROMACS executables in my github repo here. These are precompiled binaries but the redistributables for Visual C++ still has to be installed (because the binary relies on the Visual C/C++ OpenMP runtime libraries). Instructions are given in the readme file in the repository.
All of the above guide was written considering just 64-bit machines (x64). For 32-chip machines, there would exist some changes.
Source: https://medium.com/codex/compiling-and-running-gromacs-natively-on-windows-9bc40d9bc830
Posted by: royalsondritted.blogspot.com
0 Response to "How To Install Gromacs On Windows"
Post a Comment